Choose A Condition:
Please Select
» Glossary of AS measures
ASAS20 response at Week 16 (primary endpoint): 73% of patients receiving SIMPONI ARIA® achieved ASAS20 response vs 26% of patients receiving placebo (P <0.001)1,2
ASAS partial remission: Low level of disease activity was measured by criteria for “ASAS partial remission,” defined as a value below 2 on a scale of 0 to 10 in each of the 4 ASAS domains (patient global, total back pain, function [BASFI], and inflammation [average of the last 2 questions of the BASDAI concerning morning stiffness]).1
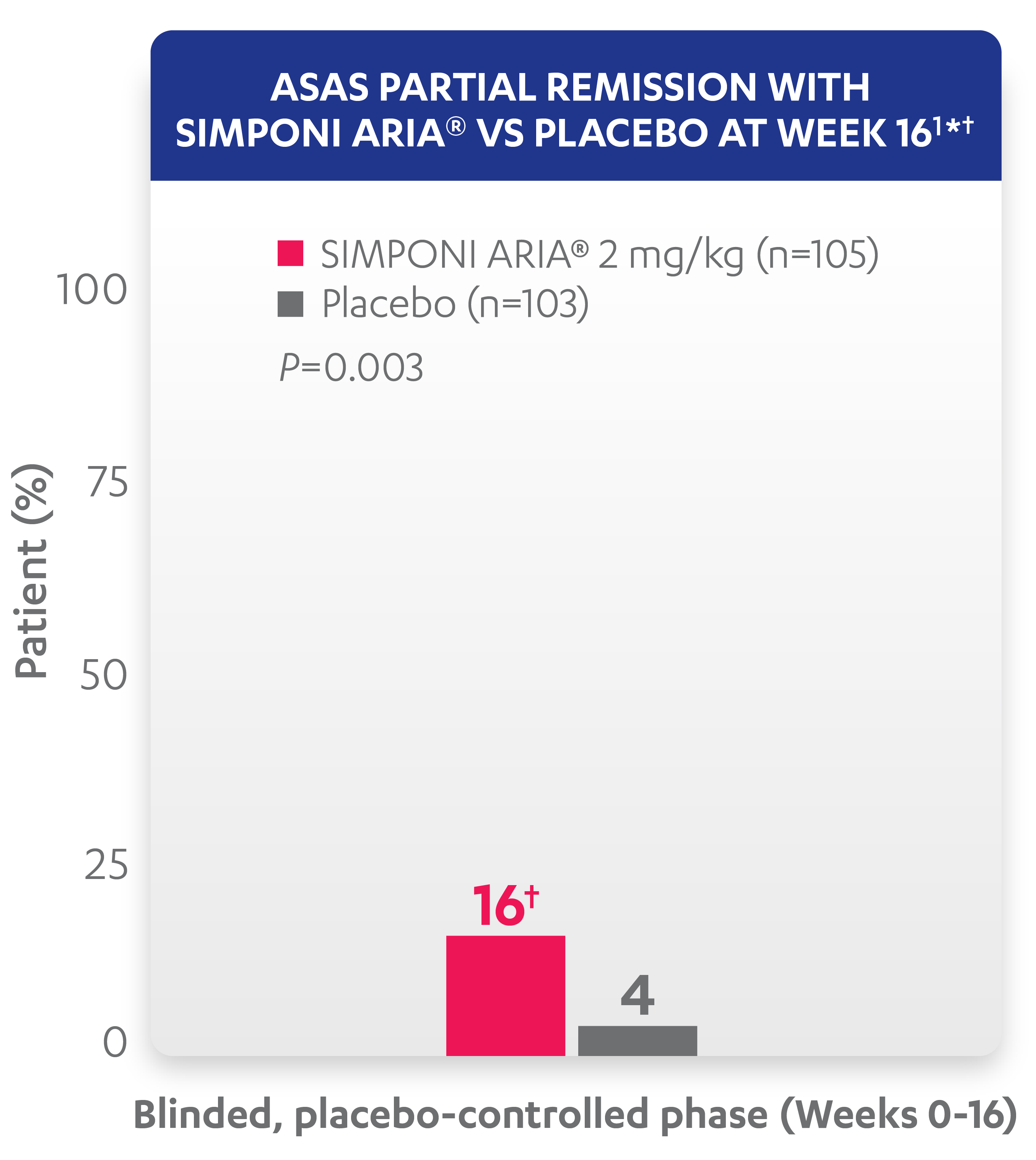
* At Week 16, all remaining patients in the placebo group began receiving SIMPONI ARIA® in a blinded manner.
† ASAS partial remission is based on imputed data using treatment failure, last observation carried forward for partially missing data, and nonresponder imputation for completely missing data.
Study design: GO-ALIVE was a global, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of SIMPONI ARIA® compared with placebo in 208 adult patients with active AS with an inadequate response or intolerance to NSAIDs. Patients had a diagnosis of definite AS for at least 3 months according to modified New York criteria. Patients had symptoms of active disease (BASDAI ≥4, VAS for total back pain of ≥4, on scales of 0 to 10 cm [0 to 100 mm], and an hsCRP level of ≥0.3 mg/dL [3 mg/L]). At Week 0, patients were randomized in a 1:1 ratio to 1 of 2 treatment groups. Subjects in the placebo group (n=103) were randomized to receive IV placebo infusions at Weeks 0, 4, and 12. At Week 16, these patients were crossed over to SIMPONI ARIA® and received administrations at Weeks 16, 20, and q8w thereafter through Week 52. Patients in the SIMPONI ARIA® group (n=105) were randomized to receive SIMPONI ARIA® 2 mg/kg infusions at Weeks 0, 4, and 12. These patients received a placebo infusion at Week 16 to maintain the treatment blind and continued to receive SIMPONI ARIA® infusions at Week 20 and q8w thereafter through Week 52. Patients were allowed to continue stable doses of concomitant MTX, SSZ, HCQ, low dose oral corticosteroids (equivalent to ≤10 mg of prednisone per day), and/or NSAIDs during the trial. The primary endpoint was the percentage of patients achieving ASAS20 response at Week 16.1
AS=ankylosing spondylitis; ASAS=Assessment of SpondyloArthritis international Society; BASDAI=Bath Ankylosing Spondylitis Disease Activity Index; BASFI=Bath Ankylosing Spondylitis Functional Index; HCQ=hydroxychloroquine; hsCRP=high-sensitivity C‑reactive protein; IV=intravenous; MTX=methotrexate; NSAID=nonsteroidal anti-inflammatory; q8w=every 8 weeks; SSZ=sulfasalazine; VAS=visual analog scale.
Reference: 1. Data on file. Johnson & Johnson. 2. SIMPONI ARIA® (golimumab) [Prescribing Information]. Horsham, PA: Johnson & Johnson.