Choose A Condition:
Please Select
FOR ADULTS WITH ACTIVE AS
» Glossary of AS measures
» Glossary of AS measures
73%

26%

37%
19%
ASAS20 response at Week 2 was not adjusted for multiplicity. Therefore, statistical significance was not established.
*An ASAS20 response is defined as an improvement of ≥20% from baseline and absolute improvement from baseline of at least 1 on a 0-to-10 scale in at least 3 of the following 4 domains: patient global, total back pain, function (BASFI), and inflammation (average of the last 2 questions of the BASDAI concerning morning stiffness). An absence of deterioration from baseline (deterioration defined as ≥20% worsening and absolute worsening of at least 1 on a 0-to-10 scale) in the potential remaining domain. An ASAS40 response is defined as a ≥40% improvement in 3 of the 4 domains with an absolute improvement of at least 2 on a 0-to-10 scale, and no worsening in the remaining domain.
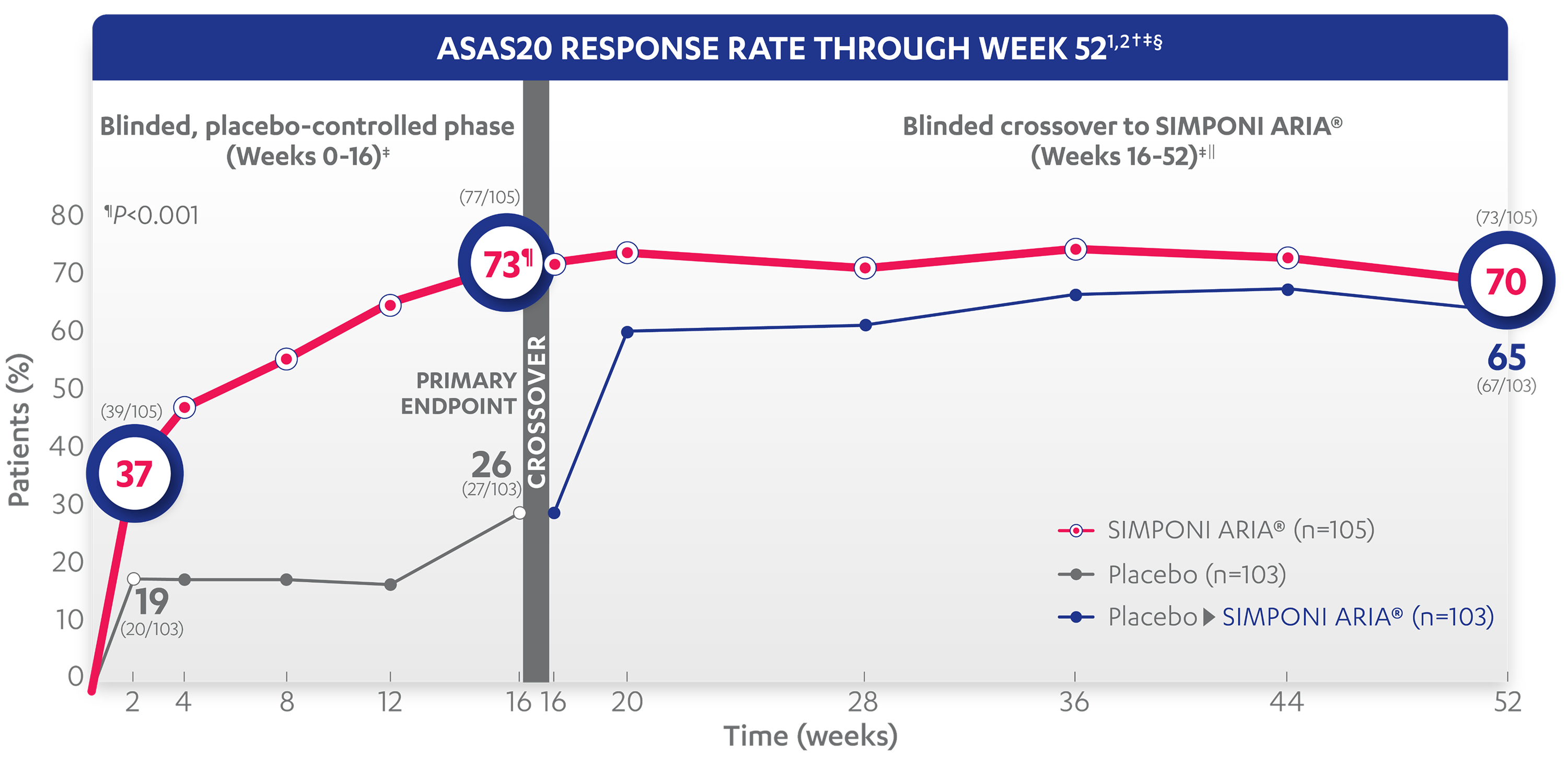
71%
62%
70%
65%
†The same patients may not have responded at each time point.
‡ASAS20 response is based on imputed data using treatment failure (only through Week 16), last observation carried forward (LOCF) for partially missing data, and NRI for completely missing data.
§In this intention-to-treat analysis, patients were considered to be nonresponders if they experienced any of the following: initiated new DMARDs, biologics, or systemic immunosuppressives; increased SSZ, MTX, or HCQ above baseline dose; initiated treatment with oral, IV, or IM corticosteroids; increased the dose of oral corticosteroids above baseline dose; or discontinued treatment due to unsatisfactory therapeutic effect. Treatment failure rules were applied only through Week 16. LOCF rules were applied for partially missing data.
||After Week 28, selected sponsor personnel were unblinded to subject-level data which may have affected results.
» Glossary of AS measures
48%

9%

14%
4%
ASAS40 response at Week 2 was not adjusted for multiplicity. Therefore, statistical significance was not established.
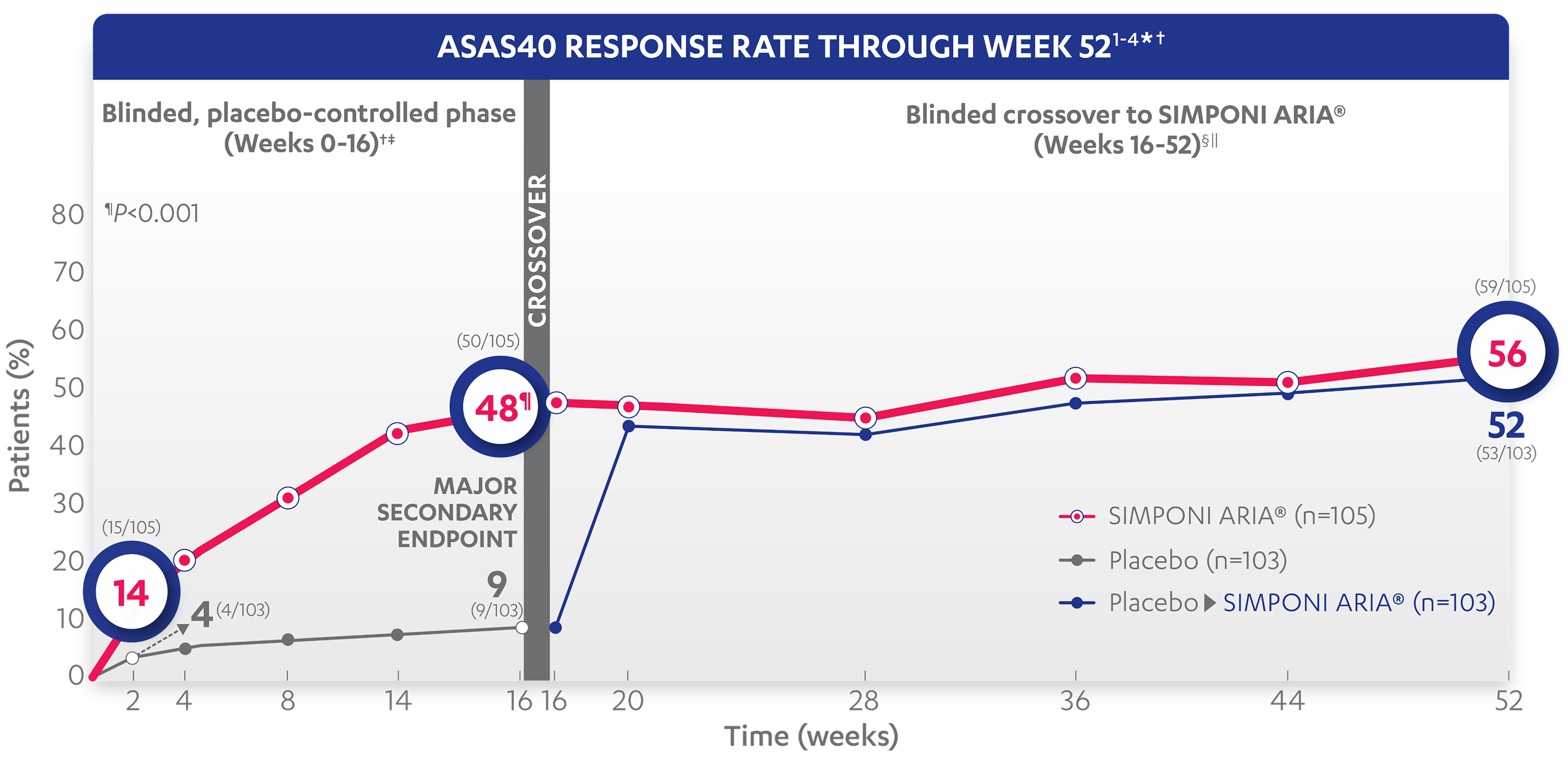
45%
43%
56%
52%
*An ASAS20 response is defined as an improvement of ≥20% from baseline and absolute improvement from baseline of at least 1 on a 0-to-10 scale in at least 3 of the following 4 domains: patient global, total back pain, function (BASFI), and inflammation (average of the last 2 questions of the BASDAI concerning morning stiffness). An absence of deterioration from baseline (deterioration defined as ≥20% worsening and absolute worsening of at least 1 on a 0-to-10 scale) in the potential remaining domain. An ASAS40 response is defined as a ≥40% improvement in 3 of the 4 domains with an absolute improvement of at least 2 on a 0-to-10 scale, and no worsening in the potential remaining domain.
†The same patients may not have responded at each time point.
‡In this intention-to-treat (ITT) analysis, patients were considered to be nonresponders if they experienced any of the following: initiated new DMARDs, biologics, or systemic immunosuppressives; increased SSZ, MTX, or HCQ above baseline dose; initiated treatment with oral, IV, or IM corticosteroids; increased the dose of oral corticosteroids above baseline dose; or discontinued treatment due to unsatisfactory therapeutic effect. Treatment failure rules were applied only through Week 16.
§Last observation carried forward (LOCF) rules were applied for partially missing data and NRI for completely missing data.
||After Week 28, selected sponsor personnel were unblinded to subject-level data which may have affected results.
References: 1. Data on file. Johnson & Johnson. 2. SIMPONI ARIA® [Prescribing Information]. Horsham, PA: Johnson & Johnson. 3. Deodhar A, Reveille JD, Harrison DD, et al. Safety and efficacy of golimumab administered intravenously in adults with ankylosing spondylitis: results through Week 28 of the GO-ALIVE study. J Rheumatol. 2018;45:3; doi:10.3899/